
Structural changes in proteins can be analyzed by molecular simulation
In other words, the human body is formed by cells that consist of proteins, and proteins are also involved in the normal function of the body.
Proteins account for about 15% of the human body. It may sound like a small part, but since 60-70% of the human body is water, almost half of the body other than water is protein.
As you know, the muscles, skin, hair, nails, organs, etc. are made of proteins, but also various enzymes, hormones, immune substances, etc. are made of proteins.
For example, when we feel sick, we may take a drug to get better. The reason why the drug works is, in short, that the drug affects cells in the body, and protein plays an important role in this process. It is a phenomenon that occurs at the atomic level between proteins and drugs.
So, if you want to create an effective drug, it is very important to look at changes in protein structure and function at the atomic level.
As you know, proteins are single stranded polymeric molecules made of amino acids linked by peptide bonds. There are twenty types of amino acids, and a combination and sequence of them determines a specific three-dimensional structure. Proteins function differently according to each structure.
Molecular simulation is one of the methods used to investigate changes in the function of proteins at the atomic level. In molecular simulations, the movement of molecules can be traced by determining the forces acting among many atoms of the protein and then solving the equation of motion numerically.
Since one amino acid consists of about 10 atoms, a protein consists of more hundreds of atoms. There are also solvents around the protein. If you treat solvent molecules in the protein system, the number of atoms in the system is more tens of thousands of atoms. The timescale for one step of the movement of atoms is measured in femtoseconds. A femtosecond is -15th power of 10 seconds, that is, 1 parts-per-quadrillion second. To investigate the dynamics of protein on this timescale, the calculated amount becomes enormous.
Actually, about 20 years ago, there were studies to reproduce the process in which water molecules were placed neatly in a low-temperature condition and crystallized, that is, it became ice, in molecular simulation. In reality, water becomes ice after several hours, however, it took about three years to simulate with a supercomputer at that time.
But back then, I was very surprised to hear that we were able to reproduce the process of water becoming crystal. There had been a discussion on whether the problem was the accuracy of the interaction or the sampling. Then, it was revealed that it was a sampling issue. Crystallization of multiple complex molecules still remains a difficult challenge.
Recently, the computational speed has been dramatically improved thanks to the supercomputer Fugaku and special-purpose hardware to accelerate molecular simulation developed in the US. Molecular simulations of cells and simulations of proteins with several hundred residues including solvents on the sub-millisecond timescale have been performed.
Even in our surroundings, the GPUs that support 3D drawing by computers have been improved dramatically, enabling us to perform molecular simulations of proteins with as many as several hundred residues on the microsecond (the -6th power of 10) timescale.
When the performance of these calculators is improved and the level of molecular simulation is raised, we can compare results with actual experimental data and obtain information about microscopic atomic movements that cannot be observed in experiments.
For example, the association between a small molecule drug and a large molecule protein can be investigated. Then, it will be applied to drug development.
Development of molecular simulation techniques
So far, we have mainly developed molecular simulation methodologies based on theories of chemical physics.
As I mentioned earlier, even with a supercomputer, it is difficult to perform a one-second simulation. For this reason, we have been developing methodologies of enhance sampling, which corresponds to a long-time simulation. This allows us to sample a variety of protein structures with a short calculation time to predict a more stable protein structure.
In addition, we are developing methods to evaluate the stability of proteins, including the effects of many water molecules around proteins, and assessing the effectiveness of these methods.
Lately, we have been working on the development of dynamic analysis methods that extract the characteristic movement of proteins from the complex movements obtained from molecular simulations of proteins.
In recent years, we have carried out the following application research using these methods.
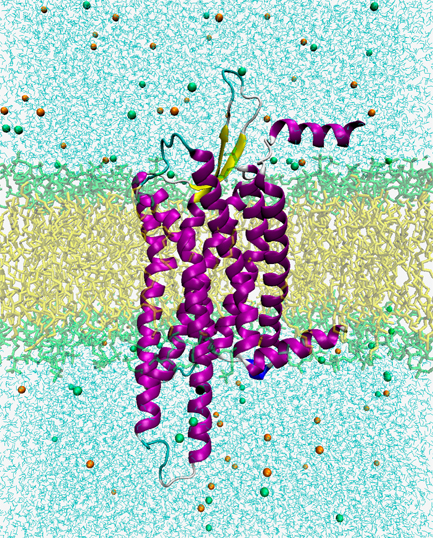
Reproducing the movement of membrane proteins affecting cells
For example, membrane proteins are embedded all over the biomembrane covering the cells. This membrane protein plays a very important role in the transmission of information inside and outside the cell and in the substance transport.
That is to say, it does not bind to everything coming to the cell, such as small molecules, ions, etc., but only to certain things. Then, membrane proteins change their structure and become bound to other proteins in the cell. This phenomenon has various effects on cells.
In other words, if you know what kind of small molecule binds to membrane protein and how the structural change of membrane protein caused by this affects cells, you can develop small molecules that change cells as you expect.
One of these membrane proteins that bind to specific small molecules and affect cells is the G protein-coupled receptor (GPCR), which has recently become a target in drug development. That means if we can send certain small molecules that trigger targeted GPCRs, it can be an effective drug.
Of course, pharmaceutical companies do not develop drugs from scratch; they have an enormous database on small molecules and then proceed with the development based on it.
Recently, the performance of X-ray crystallography and electron microscopy has been dramatically improved so that the structure of proteins and the structure of the condition of GPCRs in effect can be actually confirmed. These structures are published in the Protein Data Bank (PDB) and are available to anyone. However, these structures correspond to static images. Molecular simulations provide information on the movement of proteins at the atomic level using these structures as initial structures.
By having basic data and conducting simulations based on it, we are aiming to reproduce the movement of molecules that cannot be traced in experiments and to propose amino acid substitution which stabilizes GPCRs more or amino acid substitution which takes on an active structure more easily, because the stability of proteins can be improved, or the functions of proteins can be changed just by changing some amino acids.
In practice, simulation of membrane proteins is a complicated system since it requires setting up biological membranes such as lipids and water molecules in addition to proteins. Also, since the simulation time is only microseconds, it may be difficult to make it useful in applications, but we are progressing step by step.
Promoting basic research to help drug development
In fact, membrane proteins play an important role in immunity against cancer cells.
Unlike normal cells, cancer cells produce many foreign substances inside them. Membrane proteins present peptides, which are small fragments of the foreign substance, to the outside of cells. Then, immune system cells recognize cancer cells by detecting these peptides and attack them.
By understanding the functions of these proteins in detail, we may be able to develop some way to improve immunity against cancer cells. Again, molecular simulation should play an important role in this case.
Molecular simulation can also be utilized in vaccine developments which are conducted in various countries for the COVID-19.
As mentioned earlier, various databases are available on the Internet, including the PDB. It is also necessary for simulations to set up a system including water molecules, lipids and sugar chains as well as proteins in order to reproduce the actual biological state, and there is a website that can publish or set up these data.
For some coronaviruses, simulation setup data and trajectories are now actively published and available around the world.
During this pandemic, while social distancing is emphasized in the real world, there is a movement to promote collaboration in the online world.
In addition, in order to utilize molecular simulation in actual drug development, we need to improve its accuracy. For this purpose, it is essential to improve fundamental technologies such as supercomputers and simulation methods.
Although my own research is not yet an application research that will lead to drug development, I think that we should develop methods of simulation and apply them to individual systems, and connect them to application research as much as possible.
* The information contained herein is current as of February 2021.
* The contents of articles on Meiji.net are based on the personal ideas and opinions of the author and do not indicate the official opinion of Meiji University.
* I work to achieve SDGs related to the educational and research themes that I am currently engaged in.
Information noted in the articles and videos, such as positions and affiliations, are current at the time of production.